
Definisjon
Trombotisk trombocytopenisk purpura (TTP) er en sjelden blodsykdom. Sykdommen kjennetegnes av blodpropper (tromboser) i små blodårer og et lavt antall blodplater (trombocytopeni). Dette fører til blødninger under huden (purpura). TTP kan minne om revmatisk vaskulitt, og kan i noen tilfeller oppstå som en komplikasjon til systemisk lupus eller antifosfolipid syndrom. Utslag (lavt nivå) i blodprøven ADAMTS13 er typisk.
Sykdomsårsak
Utløsende sykdomsårsaker til TTP påvises hos ca. 50% av tilfellene. Noen kjente utløsende årsaker til TTP hos voksne er:
- Kreft
- Benmargstransplantasjon
- Svangerskap
- Medikamenter (f.eks. Acyklovir, Quinine, platehemmere, immunsupprimerende midler)
- HIV-infeksjon
- Systemisk lupus
- Antifosfolipid syndrom
- Vaksiner (f.eks. den tidligere benyttede AstraZeneca-vaksinen (Vaxzeria) mot Covid-19) (referanse: Schultz NH, 2021).
Blant voksne er årsaken er en autoimmun prosess der det dannes antistoff mot ADAMTS13 (immunologisk TTP) (referanse: Zuno JAN, 2022).
Hos barn kan TTP være forårsaket av en arvelig mangel på ADAMTS13 (kongenital/genetisk TTP). ADAM13 kan måles med redusert aktivitet hos de aller fleste. Enzymaktivitet <10% tilsvarer alvorlig form, 10-50% er uspesifikt funn og >50% er normalt. Det foreligger ikke hemmende antistoff (referanser: Fujimura Y, 2011; Upshaw-Schulman syndrom: referanse von Krogh AS, Tidsskr nor legefor, 2016; Scully M, 2021).
Ulike former for TTP
- Klassisk TTP. Når blodpropper (tromber) er hovedproblemet og nyrene er lite påvirket, kalles tilstanden klassisk TTP. Dette er den vanligste formen for TTP.
- HUS. Når nyresvikt er det dominerende symptomet, brukes ofte diagnosen hemolytisk-uremisk syndrom (HUS). Dette ses særlig hos barn med blodig diaré utløst av E. coli-bakterier.
- TTP-HUS. Det er et klinisk overlapp mellom TTP og HUS, og tilstandene omtales ofte samlet som TTP-HUS.
- HELLP. En tredje form for trombotisk mikroangiopati er HELLP-syndromet. Dette er en alvorlig svangerskapskomplikasjon.
- Idiopatisk form (kongenital /genetisk trombotisk trombocytopenisk purpura, Upshaw-Schülman syndrom/Furlan-Tsai)
- referanser: Kappler S, 2017; Zuno JAN, 2023
Symptomer
Symptomene skyldes at små blodpropper vandrer til ulike organer som hud, hjerne og nyrer (trombotisk mikroangiopati). Blodproppene kan også skade de røde blodcellene (hemolyse/hemolytisk anemi). De vanligste symptomene på TTP er:
- Nylig oppstått tretthet og utmattelse
- Hodepine (intracerebral venøs trombose, sinusvenetrombose)
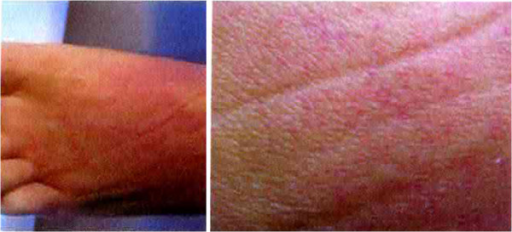
- Blødninger under huden (purpura, ekkymoser) og fra nese og tannkjøtt
- Magesmerter, oppkast, diare
- Nevrologiske symptomer som hodepine, hallusinasjoner, TIA (drypp) og hjerneslag
- I noen tilfeller nyresvikt og feber
Undersøkelser
Diagnosen TTP stilles basert på sykehistorie, kliniske funn og blodprøver.
Sykehistorien omfatter de aktuelle symptomer (se ovenfor). Ofte foreligger symptomer på infeksjon dager-uker før sykdommen bryter ut. Også symptomer på mulig bakenforliggende, assosiert sykdom bør etterspørres.
Klinisk kan det foreligge magesmerter, oppkast, diare (redusert gjennomblødning i mesenterica-arterien hos 25%), påvirket mental status, kramper, koma, nevrogene utfall (hjerne-manifestasjon hos 60%), uregelmessig puls, hjertesvikt, tung pus, frostrier og feber (hemolyse), blødninger i hud og slimhinner med purpura, ekkymoser. En grundig generell undersøkelse som også tar høyde for mulig bakenforliggende sykdom er aktuelt.
Laboratorieprøver omfatter:
- Blodplater (trombocytter). Fallende antall blodplater <30.000 x 10/l er typisk.
- ADAMTS-13-protein (<10% ved genetisk form)
- ADAMTS-13 antistoff (sendeprøve til Medisinsk biokjemi og immunologi, St Olavs Hospital, Trondheim).
- Hemoglobin (lav)
- Haptoglobin (lav ved hemolyse)
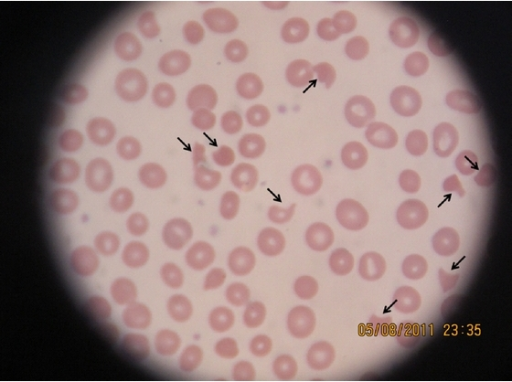
- Blodutstryk for å se etter schistocytter (fragmenterte røde blodceller); >1% i utstryk er typisk. Schistocytter forekommer også ved andre tilstander og blant friske, men i lavt antall <1% av de røde blodlegemene, mot gjennomsnittlig 8% ved TTP.
- Nyrefunksjonsprøver. Nyresvikt med økende kreatinin/fallende eGFR er sjeldnere, og en må da også vurdere om hemolytisk uremisk syndrom (HUS) foreligger (se differensialdiagnoser nedenfor).
- Andre: Direkte Coombs test og blødningstid forventes å være normale. Som screening kan CRP, SR, Hb, leukocytter med differensialtellinger, trombocytter, retikulocytter, blodutstryk: fragmenterte erytrocytter (schistocytter), lever-, nyre- og thyreoidea-funksjonsprøver, LD, glukose, urin stiks. ANA, anti-DNA, lupus antikoagulant, anti-kardiolipin og anti beta-2-glykoprotein ved henholdsvis SLE og antifosfolipid syndrom. Også HIV– og svangerskapstest vurderes etter behov. Urinprøver (stiks først) gjøres også.
Diagnosen
Diagnosen bygger på kliniske funn (symptomer og funn relatert til blodpropper og blødninger) + lavt antall blodplater (trombocytopeni) + mikroangiopatisk hemolytisk anemi uten annen åpenbar årsak. I praksis sikres diagnosen ved påvisning av et lavt ADAMTS-13 nivå eller høye antistoff.
Lignende tilstander/differensialdiagnoser
- Antifosfolipidsyndrom og CAPS: En autoimmun sykdom som fører til blodproppdannelse i både vener og arterier.
- Disseminert intravaskulær koagulasjon (DIC): En alvorlig tilstand med ukontrollert aktivering av koagulasjonssystemet som fører til både blodpropper og blødninger.
- Eklampsi (svangerskap): En alvorlig komplikasjon under svangerskapet som involverer høyt blodtrykk, kramper og ofte DIC.
- Evans syndrom (autoimmun forårsaket lavt antall blodplater og rød blodlegemer): En autoimmun sykdom som forårsaker lavt antall blodplater og røde blodceller.
- Hemolytisk uremisk syndrom (HUS og atypisk HUS): En tilstand med ødeleggelse av røde blodceller og nyresvikt, ofte utløst av infeksjon.
- Immun trombocytopeni: En autoimmun sykdom med lavt antall blodplater som fører til økt blødningstendens.
- Kreft-sykdom, paramalignt fenomen; Ulike symptomer som kan oppstå som følge av kreft, men som ikke skyldes selve svulsten.
- Malign hypertensjon: En sjelden og alvorlig form for høyt blodtrykk som kan føre til organskade.
- Nyresvikt, akutt post partum (svangerskap): Akutt nyresvikt som oppstår etter fødsel.
- PRES (lignende cerebrale MR-funn): En nevrologisk tilstand med hodepine, kramper og synsforstyrrelser, ofte utløst av høyt blodtrykk
- Renal krise ved systemisk sklerose: En alvorlig komplikasjon ved systemisk sklerose med raskt forverret nyrefunksjon.
- Shigella-infeksjon blant barn med blodig diaré: En bakteriell infeksjon som kan føre til HUS.
- Systemisk lupus (SLE), schistocytter ved immunkompleks-nedslag og vaskulitt. En autoimmun sykdom som kan påvirke mange organer, inkludert hud, ledd og nyrer.
- Vaskulitt: En betennelse i blodårene som kan føre til innsnevring eller blokkering av blodstrømmen.
Behandling
TTP er en alvorlig tilstand som ofte behandles på intensivavdeling. Tidlig behandling er viktig. Behandlingen består vanligvis av:
- Plasmaferese og infusjon av nytt plasma (plasmautskiftning) som tilførsel av ADAMTS-13.
- Kortikosteroider (SoluMedrol/Prednisolon) i høye doser ved autoimmun årsak er første linje behandling.
- Immunsupprimerende medikamenter (f. eks cyklofosfamid/Sendoxan eller biologisk behandling med rituksimab(MabThera/Rixathon).
- Caplacizumab (Cablivi) er et nyere antistoff som binder seg til von Willebrand-faktor og hindrer at blodplater klumper seg sammen. Legemidlet gis som injeksjoner under huden, og har vist seg å redusere tiden det tar for blodplatetallet å normalisere seg, samt redusere behovet for plasmaferese.
Blodtransfusjon kan utløse forverring og bør unngås. Ved medfødt form er regelmessige plasmainfusjoner med ADAMTS-13 aktuelt (referanse: Zuno JAN, 2022).
Prognose
Tilstanden er alvorlig i den akutte fasen. Selv med behandling er det en risiko for tilbakefall (residiv). Tilbakefall ses ofte etter 1-2 år, men kan også forekomme etter 20 år. Gjentatte behandlinger med rituksimab kan redusere risikoen for tilbakefall (referanse: Zuno JAN, 2022).
Litteratur
- Zuno JAN, 2023
- Dane K, 2018
- Kremer Hovinga JA, 2017
- Ben-Amor A, 2015
- von Krogh AS,Tidsskr nor legefor, 2016 (Kongenital form: Kongenital trombotisk trombocytopenisk purpura)
- Grans revmakompendium